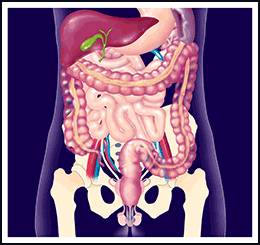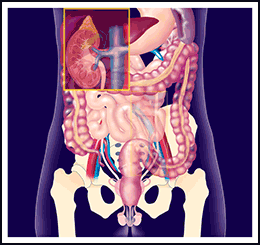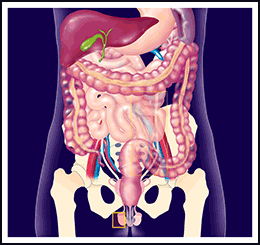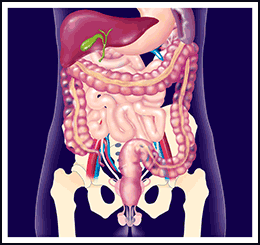
Che cosa è il rene policistico dell’adulto?
Nome Inglese: Autosomal Dominant Polycystic Kkidney Disease (ADPKD)
Il rene policistico dell’adulto, che si indica anche con la sigla ADPKD, è una delle malattie genetiche più comuni. La sua incidenza è calcolata essere pari a 1 su 1000 ed è la principale causa genetica di insufficienza renale dell’adulto.
La caratteristica principale di questa malattia è il formarsi di cisti in entrambi i reni.
Le cisti aumentano sia nel numero che nelle dimensioni durante la vita di un individuo, fino a causarne la perdita totale della funzionalità renale in circa la metà dei pazienti.
Questa malattia è tuttavia "sistemica" perchè altri organi oltre al rene ne possono essere colpiti.
Talvolta infatti sono presenti cisti anche in altri organi limitrofi (fegato e/o pancreas).
Il sistema cardiovascolare può anche essere interessato in questa patologia con l’insorgere di ipertensione e di aneurismi.
Quando si manifestano i primi sintomi del rene policistico?
Si tratta di una malattia ad esordio tardivo: in genere i primi segni clinici, rappresentati da dolori lombari, micro o macroematuria (presenza di sangue nelle urine), comparsa di ipertensione arteriosa, si manifestano tra i 40 e i 50 anni di età.
L’età d’insorgenza della malattia, la gravità e il decorso clinico sono tuttavia molto variabili: si ritiene che siano condizionati oltre che da fattori genetici anche da fattori ambientali.
Quali sono le cause genetiche del rene policistico?
L’ADPKD è una malattia geneticamente eterogenea. Può essere causata dalle mutazioni in due geni, denominati PKD1 e PKD2, che possono provocare forme della malattia non distinguibili fra loro, anche se sembra che la forma ADPKD2 sia leggermente meno grave rispetto ad ADPKD1.
Il gene PKD1, mutato in circa l’85% dei pazienti, si trova sul cromosoma 16.
Il gene PKD2, localizzato sul cromosoma 4, risulta mutato nel rimanente 15% circa dei pazienti.
Quali sono i possibili approcci terapeutici?
La malattia (Autosomal Dominant Polycystic Kidney Disease, ADPKD), è la più comune forma di malattia renale cistica e rappresenta, nel mondo, la causa di terapia sostitutiva emodialitica nel 7-10% dei pazienti. Sono noti due tipi di malattia policistica: il tipo I è causato da mutazioni del gene PKD1, che codifica per la policistina-1, è la forma più diffusa ed aggressiva e colpisce soggetti di età giovane; il tipo II è causato da mutazioni del gene PKD2 che codifica per la policistina-2 e rappresenta il 10-15% dei casi, ad evoluzione più lenta.
Clinicamente, le cisti si sviluppano a livello renale, epatico, pancreatico ed intestinale.
Il dolore cronico, la chirurgia palliativa, l’insufficienza renale, la dialisi, il trapianto, come anche la morte, sono tutte conseguenze di questa malattia genetica che non ha ancora una terapia medica per rallentare o arrestare la sua progressione.
Di grande interesse per il suo potenziale terapeutico, è la dimostrazione che la Policistina-1, formando un complesso con la tuberina (la proteina la cui mutazione causa la sclerosi tuberosa), agisce come un inibitore endogeno dell’attività del mammalian Target of Rapamycin (mTOR).
Se mutato, come nell’ADPKD, tale meccanismo inibitorio viene compromesso e ciò favorirebbe lo sviluppo delle cisti.
I recenti discordanti risultati di alcuni studi nell’uomo sull’uso di un inibitore di mTOR in pazienti affetti da ADPKD, possono generare interrogativi e confusione, ma diverse e molteplici possono essere le ragioni per cui tali studi hanno portato a conclusioni diverse fra di loro.
A questo punto, è d’obbligo porsi l’ interrogativo se questi risultati siano la fine o possano essere l’inizio di nuovi studi.
Agli Autori piace considerare la seconda ipotesi, in quanto tutti gli studi di biologia molecolare, quelli preclinici, e su animali, hanno confermato la “bontà” del percorso intrapreso.
Vi consigliamo di visitare anche il sito internet seguente:![]()